

 Four Years, Five Stages: the FDA's Final Rule on LDTs">
Four Years, Five Stages: the FDA's Final Rule on LDTs">
After over a decade-long saga, the Food and Drug Administration (FDA) published its final rule on Laboratory Developed Tests (LDTs) on May 6, 2024, with an effective date of July 5, 2024. As repercussions to the publication of this final rule emerge over the next several months, health systems, reference and commercial laboratories, diagnostic and reagent manufacturers, regulatory and compliance organizations, patients and their advocacy groups will debate the balance between patient safety and quality and innovation which drives cutting edge and potentially life-saving diagnostics. We share today our view of the final rule and the downstream implications for complex molecular diagnostic testing including clinical Next Generation Testing (NGS) offered as LDTs.
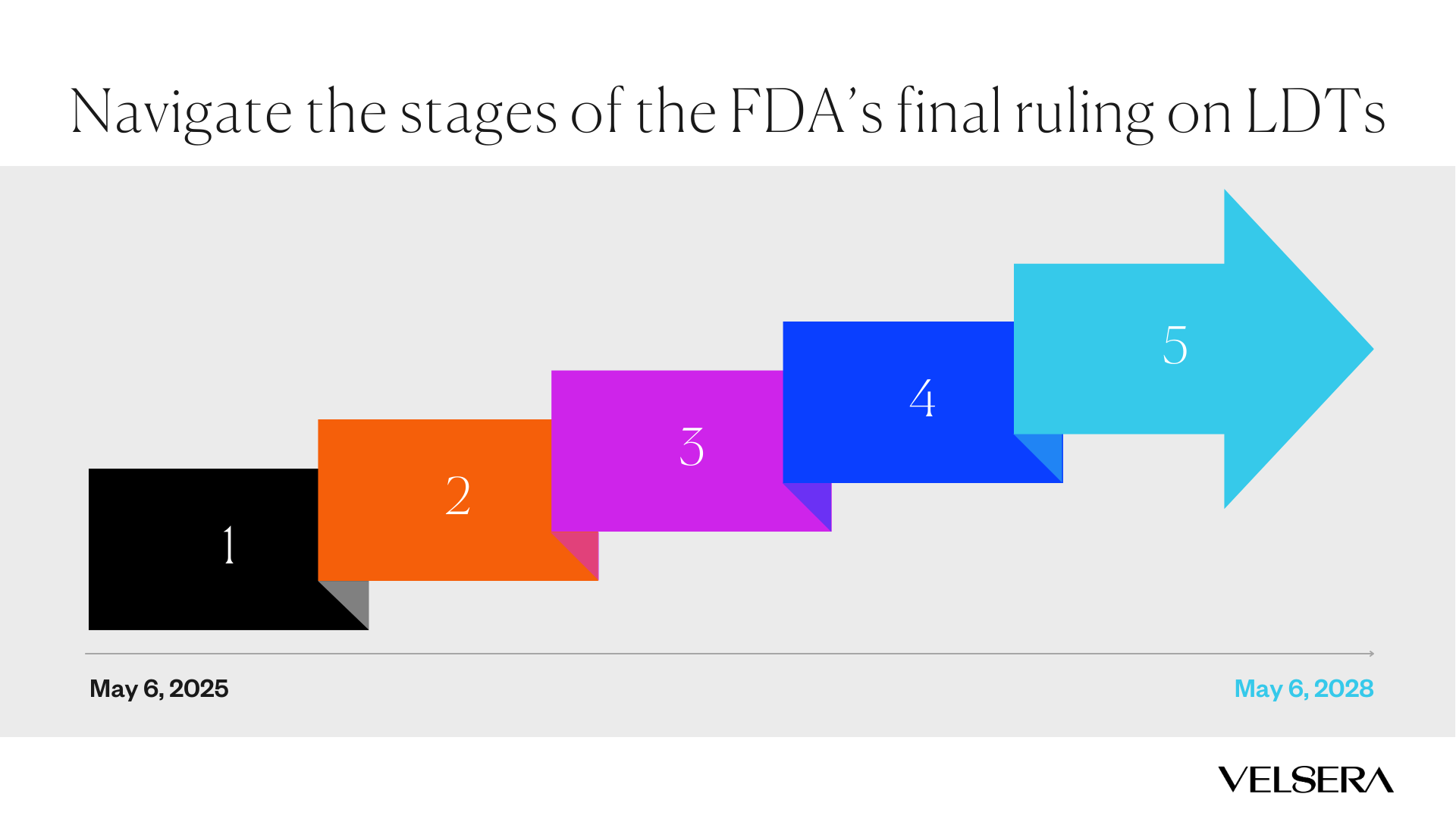
With this ruling, the FDA updated its definition of an in vitro diagnostic product (IVD) to include tests that are manufactured by individual laboratories. Prior to this ruling, the FDA exercised enforcement discretion for individual diagnostic laboratories running LDTs and reserved regulatory oversight for commercial assay manufacturers. The FDA will phase out this enforcement discretion policy over a period of four years with five stages.
.png?width=1920&height=1080&name=FDA%20stages%20(4).png)
Stage 1:
Beginning on May 6, 2025, laboratories will need to comply with medical device reporting (MDR), correction and removal reporting, and complaint file requirements. This means being able to track, investigate, and report any adverse events, product recalls, and customer complaints related to their test with robust procedures and systems.
Stage 2:
Beginning on May 6, 2026, laboratories must comply with new requirements for establishment registration, device listing, labeling, and investigational use of their LDTs.
Stage 3:
Beginning on May 6, 2027, laboratories must have a compliant quality system in place for all remaining processes. This includes implementing processes such as auditing, design controls, risk management, and management responsibility outlined in the Quality Management System Regulation (QMSR).
Stage 4:
Beginning on November 6, 2027, laboratories must comply with premarket review requirements for high-risk (class III) IVDs offered as LDTs. A lab that submits a premarket review application by this date is also considered in compliance pending completion of FDA review.
Stage 5:
Beginning on May 6, 2028, laboratories must comply with premarket review requirements for moderate-risk and low-risk IVDs offered as LDTs. A lab that submits a premarket review application by this date is also considered in compliance pending completion of FDA review.
However, there are several scenarios where the FDA will continue to exercise enforcement discretion to some degree. These include:
- LDTs for which an FDA-approved IVD does not exist and the test is performed in an integrated healthcare system. These tests will be exempt from most Quality System Regulation (QSR) requirements and premarket review requirements only until an FDA-approved IVD is available. For example, clinical NGS testing of rare diseases for which no FDA-approved or cleared IVD exists would be exempt.
- Laboratories whose tests have been approved or conditionally approved by New York State’s Clinical Laboratory Evaluation Program (CLEP) will be exempt from premarket review requirements.
- LDTs that are run by laboratories at the Department of Defense and Veterans Affairs medical facilities are exempt from all requirements.
Practically, what does this mean for laboratories that are marketing PCR or NGS-based LDTs? While not meant to be a comprehensive list of considerations, such laboratories should:
- For health system affiliated laboratories, assess each current LDT offering to determine if it meets an unmet need
- For all laboratories, assess whether adopting an existing FDA-authorized IVD is reasonable and appropriate.
- For all laboratories, where at least one LDT will fall under FDA oversight:
-
- Assess whether the test is high- versus moderate- or low-risk.
- Assess whether New York State CLEP submission is applicable or whether utilization of FDA’s Third Party review program is appropriate to gain FDA authorization.
- Perform gap assessments between their current policies, practices, and compliance under CLIA or a deeming authority under CLIA and FDA’s requirements.
- Develop a detailed plan for each marketed product by stage to be compliant under the final rule’s phase-out timelines.
-
While the next months and years are likely to result in major changes to the molecular diagnostic laboratory operations nationally, Velsera is ready to support our current and future partners on this journey. We will bring our decades-long experience in supporting complex molecular diagnostic testing, meeting regulatory compliance under CLIA and by the FDA, supporting the validation and determination of the performance of LDTs, and providing clinical-grade software solutions in support of our combined mission to offer safe and cutting edge complex molecular diagnostic tests in a democratized fashion nationally and globally.
Subscribe to our newsletter and follow us on social media for more updates as we delve deeper into each component of the FDA’s final rule in the coming weeks and months. We appreciate your partnership as you navigate your Precision Medicine initiatives.


